日期:2026-05-27 浏览人数:313人
2026年5月,美国FDA正式终结实施长达32年的DSHEA宽松监管体系或将取消“备案即可销售”的宽松模式,全面推向“强制列名、原料严格审查”的强监管时代。
这场被业内称为“从放任到严管”的变革,彻底颠覆了沿用近30年的行业规则,不仅将重塑全球保健品市场格局,更给从事美国跨境进出口的商家带来了深远影响。

早在1994年,美国国会通过DSHEA法案,赋予FDA监管膳食补充剂的权力,但未强制要求企业将产品实际注册。
但是,在此后的32年间,美国保健品数量从4000种激增至10万种以上,年销售额从40亿美元飙升至500亿美元,超过四分之三的美国成年人服用膳食补充剂。
然而,伴随市场野蛮生长的是严重的安全隐患。由于不良反应严重漏报,FDA估计实际年不良事件超过5万起。为了消除监管盲区,2026年5月,FDA正式终结这一宽松体系,推动保健品行业走向规范化发展。
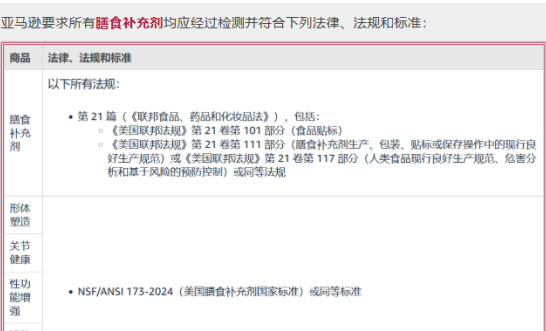
其实,在膳食补充剂方面,亚马逊有严格的要求,不仅需要卖家需要符合在膳食补充剂的要求,而且在产品页方面也有相关的规定。

目前,本次改革法案尚在国会推进,还未正式生效。不过业内人士认为,彻底推翻沿用32年的行业旧规。

根据全新《膳食补充剂列名法案》,所有在美销售的膳食补充剂,其生产、包装、经销企业必须向FDA完成全维度备案,申领唯一官方列名编号。无备案编号产品,一律按假货、错标产品处理,禁止全美销售流通。
FDA将NDI新型原料审核列为2026年核心工作。针对1994年后上市的新兴原料,企业必须提交完整的原料资质、安全实验数据及功效佐证材料,经FDA严格审核通过后方可使用。
同时FDA上线AI智能审查系统,精准甄别无安全依据的概念原料、虚假原料,严厉打击行业原料炒作、造假乱象。
沿用32年的企业GRAS自我认证模式全面取消。新规明确,所有宣称“一般安全(GRAS)”的膳食成分,不再由企业自主判定安全,必须提交官方备案及安全材料,由FDA统一审核认定,从源头杜绝安全造假。
本次新政由美国参众两院联合推动,确立“先备案、后上市”硬性规则,覆盖维生素、益生菌、草本提取物等全品类膳食补充剂。预计所有入市产品必须完整提交9大类备案信息:
1. 产品识别信息:产品名称、品牌名称、口味等所有变体信息;
2. 责任方信息:企业全称、详细地址、负责人及运营人联系方式;境外企业必须额外提供美国境内代理人信息、境内地址及官方联系方式;
3. 完整标签资料:产品全套标签高清电子版;
4. 全成分明细:包含专有配方内所有成分、每份精准含量(核心配比受法律保密保护,无需公开披露);
5. 营养使用说明:食用份量、单容器份数、营养素参考值、官方使用指南;
6. 安全警示声明:过敏原标注、风险警告、安全储存及使用规范;
7. 功效宣称内容:包装、说明书上所有结构功能、健康关联、疾病预防相关宣称内容;
8. 产品形态信息:片剂、胶囊、粉末、液体、软糖等具体产品形态;
9. 官方唯一编号:备案审核通过后,获取FDA专属列名编号,必须印刷在产品标签上,作为合法入市凭证。
中国是美国保健品核心原料供应国与OEM代工基地,多数企业长期依赖监管灰色地带,混淆“FDA销售资质”与“功效认证”,以低价模式抢占市场。本次最强监管升级彻底压缩灰色空间,行业迎来全面合规洗牌。
对中国跨境卖家、原料厂商、OEM企业的核心影响如下:
1. 合规门槛暴涨:原料审核、配方备案、标签规范、供应链溯源全流程标准化,无合规能力的中小商家加速出局;
2. 告别自证红利:成分安全不再可自主申报,所有配方、原料需官方审核,新品上市周期大幅拉长;
3. 供应链成核心壁垒:完整的溯源文件、合规台账为运营刚需,文件管理混乱、溯源缺失将直接导致退市;
4. 行业格局重塑:低价无序竞争落幕,合规优质企业抢占市场,行业走向品牌化、规范化。
除膳食补充剂外,以下出口美国的品类均需严格遵守FDA监管标准,跨境商家可自查合规:
加工/冷冻/包装食品、饮料、膳食补充剂;各类食品接触包装、厨具餐具等,均需符合FDA安全标准。
处方药、非处方药、原料药、疫苗等生物制品,生产、包装、标签、上市全流程需FDA审核监管。
口罩、防护设备、注射器、医美植入物、助听器等设备,按风险等级划分三类审核标准,分级合规。
护肤品、清洁产品、彩妆、香水、染发剂、牙膏等,需满足成分与标签规范,企业及产品需完成注册列名。
激光辐射设备、兽药、宠物食品、养殖饲料、烟草制品
欧代易:为您提供欧盟FDA认证一站式服务
欧代易深耕欧盟合规领域多年,致力于为跨境卖家提供专业、高效的合规方案,一站,助您无忧出海!若您有欧盟EPR注册或检测的需求,欢迎随时联系欧代易客户经理,或扫描下方二维码在线咨询!






